近日,中國科學(xué)院合肥物質(zhì)院健康所劉青松藥學(xué)團隊研發(fā)出一種新型受體相互作用蛋白激酶1(RIPK1)抑制劑,為程序性壞死(necroptosis)相關(guān)炎癥性疾病的治療提供了新的候選藥物。該研究成果發(fā)表于藥物化學(xué)領(lǐng)域權(quán)威期刊Journal of Medicinal Chemistry。
研究表明,炎癥性腸病(IBD)和全身性炎癥反應(yīng)綜合征(SIRS)等炎癥性疾病的發(fā)病機制與程序性壞死密切相關(guān)。當(dāng)細胞發(fā)生程序性壞死時,細胞膜破裂并釋放出大量細胞損傷相關(guān)物質(zhì),誘發(fā)炎癥反應(yīng)。RIPK1-RIPK3-MLKL信號軸被認(rèn)為是程序性壞死的核心通路,其中RIPK1是該通路的關(guān)鍵調(diào)節(jié)蛋白。因此,抑制RIPK1的激酶活性成為治療相關(guān)炎癥性疾病的潛在策略。
研究團隊前期通過藥物重定位策略,發(fā)現(xiàn)處于臨床階段的FGFR抑制劑AZD4547能夠通過抑制RIPK1發(fā)揮抗程序性壞死作用,并在小鼠SIRS模型中驗證了其治療效果(Acta Pharmacologica Sinica, 2023, 44(4):801-810)。在此基礎(chǔ)上,研究人員以AZD4547的骨架為起點,結(jié)合計算機輔助藥物設(shè)計和Ⅱ型激酶抑制劑設(shè)計策略,通過系統(tǒng)的構(gòu)效關(guān)系研究和結(jié)構(gòu)優(yōu)化,獲得了新型1H-吡唑-3-胺衍生物44。在體外活性測試中,該化合物具有顯著的RIPK1抑制活性(IC50 = 5.33 nM),并在219種蛋白激酶的廣譜篩選中表現(xiàn)出優(yōu)異的選擇性。在TSZ誘導(dǎo)的人類及小鼠細胞模型中,化合物44可以有效阻斷程序性壞死。此外,該化合物以劑量依賴方式抑制RIPK1及其下游RIPK3和MLKL的磷酸化。藥代動力學(xué)研究表明,化合物44在大鼠和小鼠上具有良好的口服生物利用度,且展現(xiàn)出較高的血液暴露量。進一步體內(nèi)研究發(fā)現(xiàn),在TNF-α誘導(dǎo)的SIRS模型中,口服化合物44(10 mg/kg)能有效阻止小鼠體溫下降并提高存活率;在DSS誘導(dǎo)的IBD模型中,其可以有效減輕結(jié)腸縮短和炎癥損傷。綜上所述,化合物44作為一種高活性、高選擇性且具有良好成藥性的新型RIPK1抑制劑,展現(xiàn)出顯著的抗炎潛力,是具有進一步開發(fā)價值的候選藥物。
該研究工作獲得了國家自然科學(xué)基金、安徽省科技重大專項和安徽省重點研發(fā)計劃等項目的支持。
文章鏈接:https://doi.org/10.1021/acs.jmedchem.5c02124
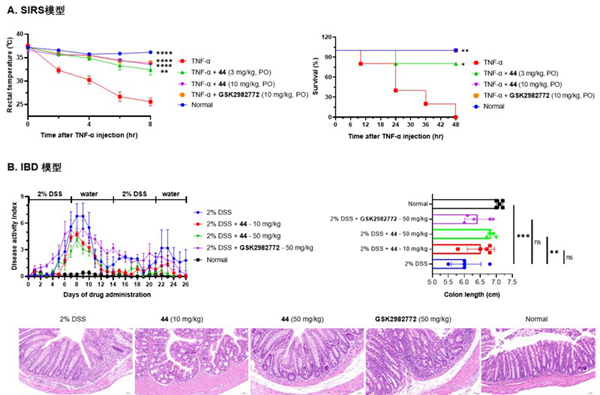
化合物44在多種炎癥性疾病動物模型中的治療效果

<span id="9mlez"><optgroup id="9mlez"></optgroup></span>



