近日,中國科學院合肥物質院健康所劉青松藥學團隊研發出一種新型嘌呤類PGK1抑制劑,該化合物在炎癥性腸病(IBD)模型中表現出良好的治療效果。相關研究成果在線發表于藥物化學領域權威期刊《Journal of Medicinal Chemistry》。
IBD是一種慢性、復發性腸道炎癥疾病,嚴重影響患者的生活質量。現有治療藥物如糖皮質激素、環孢菌素A、烏帕替尼等均伴隨不可忽視的副作用。磷酸甘油酸激酶1(PGK1)作為糖酵解過程中的關鍵代謝酶,近年來被發現是IBD的潛在治療靶點。然而,現有PGK1抑制劑在活性或藥代動力學性質方面的局限性限制了它們的臨床應用。因此,發展具有新型骨架及良好成藥性的PGK1抑制劑具有重要意義。
本研究在課題組前期通過高通量篩選發現的苗頭化合物NG52的基礎上,采用基于結構的理性藥物設計,系統地對嘌呤衍生物NG52的R1、R2和R3區域開展構效關系研究,合成了一系列新化合物,并檢測了它們對PGK1的抑制活性。經過多輪結構優化,發現嘌呤衍生物6e不僅在酶活水平上表現出優異活性(IC50),而且在大鼠和小鼠中均展現出良好的生物利用度(F)和較高的口服暴露量(AUC)。此外,在激酶選擇性評估中,6e在210種激酶中對PGK1具有高選擇性。進一步體外研究顯示,6e顯著抑制PGK1介導的糖酵解過程,降低葡萄糖消耗和乳酸生成,上調Nrf2蛋白,促進HO-1蛋白表達,抑制IL-1β和IL-6的轉錄與蛋白水平,從而發揮抗炎作用。在動物結腸炎模型中,口服6e(10和30 mg/kg)能夠劑量依賴性地緩解體重下降、降低疾病活動指數(DAI)、改善結腸縮短和組織病理損傷,顯示出良好的體內抗炎療效。綜上所述,該研究為PGK1靶向治療IBD提供了具有潛力的臨床前候選藥物。
本研究工作獲得了國家自然科學基金、安徽省科技重大專項和安徽省重點研發計劃等項目的支持。
文章鏈接:https://pubs.acs.org/doi/10.1021/acs.jmedchem.5c01334
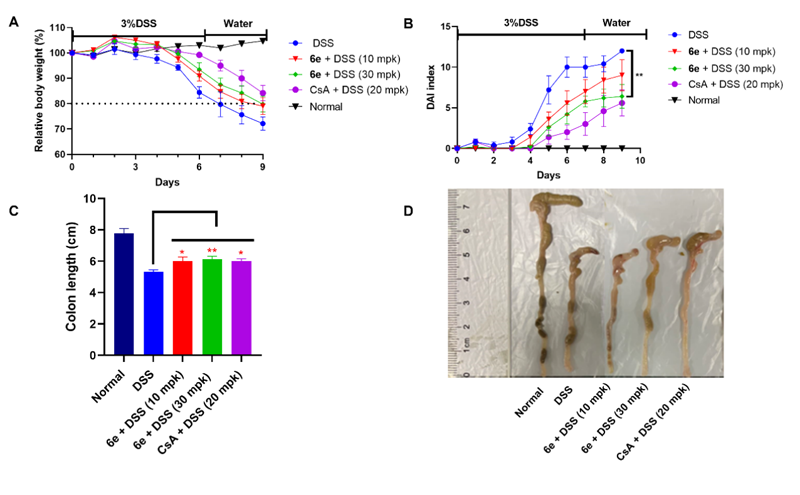
化合物6e在DSS誘導的小鼠急性結腸炎模型中的治療效果

<span id="9mlez"><optgroup id="9mlez"></optgroup></span>



